| Paciente afecta de enfermedad de Fabry | |||||
Mari Cruz Sánchez García |
Servicio Diálisis y Trasplante Renal. |
|||||
La enfermedad de Fabry fue descrita por primera vez en 1898 y cien años más tarde se estableció toda la secuencia genética. Debe su nombre a los dermatólogos J. Fabry y W. Anderson. Es un trastorno hereditario debido a mutaciones de la región Xq22.1 del cromosoma X, lo que ocasiona una ausencia o disminución de la actividad de la alfa-galactosidasa A (a-Gal A), una enzima lisosomal, lo que ocasiona una progresiva acumulación de glucoesfingolípidos neutros, principalmente la globotriaosilceramida (GL3), en las paredes de los vasos sanguíneos y musculatura lisa, provocando un importante compromiso multiorgánico. Los órganos con mayor afectación son: riñones, corazón, sistema nervioso y piel, pudiendo causar insuficiencia renal, hipertrofia ventricular, arritmias cardíacas o embolias cerebrales, reduciendo por ello la esperanza y la calidad de vida 1-4. Aunque está catalogada como Enfermedad Rara ya que su incidencia es baja: (1/ 476.000 habitantes), es la segunda enfermedad más frecuente de los 40 trastornos genéticos descritos por depósito lisosomal4. Las enfermedades lisosomales son un grupo de más de 45 trastornos genéticos diferentes, todos ellos debidos a una disfunción lisosomal que conlleva una anormal acumulación en el interior celular de sustancias que en condiciones normales son degradadas. Se presentan de manera infrecuente y a diferencia de otros grupos de enfermedades metabólicas suelen alterar su fenotipo a lo largo de su evolución, por lo que su sintomatología va variando5,6.
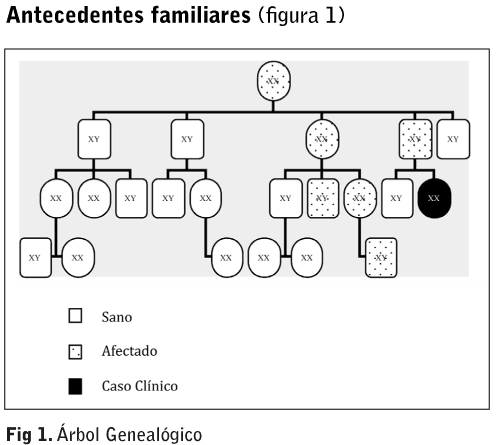
La mutación que se produce es autosómica dominante ligada al cromosoma X, aunque pueden existir casos esporádicos. La enfermedad se hereda ligada al cromosoma X por lo que uno o ambos padres pueden ser portadores de este gen mutante. Ha sido descrita mayoritariamente en hombres, pero también existen casos diagnosticados en mujeres. Los hombres con su único cromosoma X afectado (hemizigotos) sufren la enfermedad, mientras que las mujeres que heredan un único cromosoma X afecto (heterozigotas) tienen generalmente síntomas más leves. La transmisión de la enfermedad se produce de la siguiente manera: en cada célula se inactivan al azar uno de los 2 cromosomas y es el otro el que aporta la dotación genética. Por ello, los varones hemizigotos tendrán todos sus hijos varones sanos siendo sus hijas portadoras de dicha enfermedad. Las mujeres heterozigotas tendrán un 50% de probabilidad de tener tanto varones hemizigotos como mujeres heterozigotas, ya que heredan un cromosoma X de cada padre7. Las manifestaciones clínicas son variadas7-9: Renales: el depósito de Gb3 en las células epiteliales y endoteliales de la cápsula de Bowman y en las células epiteliales de los túbulos contorneados distales del Asa de Henle provoca proteinuria y disminución de la función renal. Cardíacas: miocardiopatías, hipertrofia ventricular izquierda y arritmias. Neurológicas: en el sistema nervioso periférico se produce una neuropatía, provocando hipohidrosis (disminución o ausencia de la sudoración) e intolerancia al calor. Todo ello debido a la acumulación de Gb3 en las glándulas sudoríparas. En el sistema nervioso central pude provocar ictus, hipertensión, epilepsia, afasias, convulsiones, alteraciones psiquiátricas/ cognitivas como depresión o ideas autolíticas. Cutáneas: angioqueratomas que son lesiones máculo papulares de color rojizo-purpúreo que no se blanquean con la presión y suelen agruparse en forma de racimo. Pueden aparecer en muslos, nalgas, ombligo, zona escrotal y pene llegando a persistir de forma indefinida. Oculares: córnea verticillata que se produce una queropatía en forma de remolino debido al depósito del Gb3 en el epitelio corneal, provocando un color grisáceo-dorado del epitelio corneal inferior y líneas arborizantes que se inician por debajo de la pupila y se dirigen hacia la periferia simulando los radios de una bicicleta. También pueden producirse trastornos vasculares y catarata de Fabry. Digestivas: diarreas, dolor abdominal. Existe una triada fenotíca característica: dolor neuropático distal, angioqueratomas e hipohidrosis, aunque ésta es una enfermedad progresiva que suele iniciarse con acroparestesias constantes o crisis de Fabry (dolor agudo con sensación de quemazón y escozor que puede irradiarse a otras zonas del cuerpo) posteriormente aparecen los angioqueratomas y la hipohidrosis. Muchos de sus signos y síntomas comienzan durante la infancia o adolescencia. Si no se instaura tratamiento la enfermedad progresa hacia el compromiso renal y cardiovascular8,9. El diagnostico de Fabry se confirma mediante pruebas de laboratorio: en caso de sospecha de la enfermedad en hombres, bastará con la detección en sangre (leucos/ plasma) de una concentración baja de la enzima- Gal A. Para las mujeres resulta mucho más eficaz la confirmación genética mediante análisis mutacional del Gen GAL en el ADN. Por el contrario, si ya existe un caso índice en la familia, es necesario realizar un estudio genético siempre, tanto en hombres como en mujeres. También es necesario la elaboración de un árbol genealógico para averiguar qué otros familiares pueden estar afectados, y quienes pueden estarlo en el futuro4. Hasta hace pocos años, sólo podía realizarse tratamiento sintomático o paliativo que se centraba en tratar los síntomas básicos de la enfermedad. Para el dolor se utilizaban antiinflamatorios, los angioqueratomas se trataban con láser de argón o vapor y se administraban anticoagulantes y antiagregantes para la sintomatología cardiovascular. En cuanto a la enfermedad renal era necesario en la mayoría de casos iniciar tratamiento renal sustitutivo además de los inhibidores de la angiotensina. Para aliviar los síntomas digestivos debían llevar una dieta pobre en grasas y en algunos casos era necesario la metoclopramida3. Desde el 2001 existe un tratamiento para la enfermedad que se basa en el reemplazo enzimático. En Europa existen 2 formas comercializadas de la enzima alfa galactosidasa: agalsidasa alfa (Replagal®) que se obtiene por ingeniería genética a partir de células humanas; y la agalsidasa beta (Fabrazyme®), que se obtiene a partir de células de ovario de hámster chino. La dosis a administrar varía en función del peso y de la opción farmacéutica que se escoja: 0,2 mg/kg para el Replagal® y 1 mg/kg para el Fabrazyme ®. Ambos fármacos intervienen en el control del almacenamiento de los glucoesfingolípidos4,10. El tratamiento se realiza de forma ambulatoria en nuestro servicio de hemodiálisis y trasplante renal. Se debe acudir cada 15 días a recibir la dosis de agalsidasa alfa. Ésta se prepara en el servicio de farmacia de nuestro hospital. Para la administración endovenosa de la medicación (10,5 mg) es necesario instaurar un acceso vascular de calibre pequeño (G20, G21). La dosis de la enzima viene diluida en 100 ml de suero fisiológico y la administración se realiza entre 40 y 60 minutos. Se proceden a controles hemodinámicos de TA, FC y Tª antes y después del tratamiento. Al finalizar la infusión se vuelve a purgar el equipo para administrar la dosis en su totalidad. Presentamos un caso clínico en el que se describe la sintomatología presentada por la paciente y su evolución tras recibir tratamiento de reemplazo enzimático con Replegal® durante más de cinco años. CASO CLÍNICO Se trata de una mujer de 33 años heterozigota, que a los 27 años se le diagnostica la enfermedad de Fabry desde el punto de vista enzimático y molecular: mutación W815 situada en el exón 2 del Gen GAL en la región Xq 22.1 del cromosoma X. En el momento de su diagnóstico la paciente presentaba la siguiente sintomatología:
• Padre: fallecido. Presentaba asma bronquial desde los 20 años, hipohidrosis, cardiopatía en forma de hipertrofia miocárdica severa, acroparestesias, cólicos abdominales, fiebres de origen desconocido, angioqueratomas en testículos y zona anal, desarrolló IRC y precisó programa de hemodiálisis. También presentaba demencia y desorientación debido a una afectación neurológica central y afectación vascular, por lo que requirió ingreso en institución psico-geriátrica dado la imposibilidad de un buen control por parte de la familia debido al estado avanzado de su enfermedad. Recibió tratamiento de reemplazo enzimático. • Abuela materna: se desconoce tipo de afectación. Dados sus antecedentes familiares, se le realizaron las pruebas médicas pertinentes diagnosticándosele en noviembre de 2003 la enfermedad de Fabry. La paciente dio el consentimiento verbal para ser incluido en el FOS (Fabry Outcome Survey) un registro internacional independiente y multicéntrico donde se incluyen a los pacientes que están recibiendo tratamiento de reemplazo enzimático, o que en un futuro pueden llegar a recibirlo, por lo que representa la base de datos más completa sobre la evolución clínica de enfermos afectados por Fabry a nivel internacional. Los nefrólogos junto con pediatría le propone iniciar tratamiento sustitutivo enzimático, la paciente acepta el tratamiento y la 1ª dosis se administra el 31 octubre 2003. La paciente sigue los controles por los siguientes especialistas: digestivo, cardiología, oftalmología, neurología, dermatología y nefrología, ya que para el buen control de la enfermedad es necesaria la actuación conjunta de todas estas disciplinas. Dada su sintomatología en el momento del diagnóstico se establecieron los siguientes diagnósticos enfermeros: Desde el inicio del tratamiento no se han producido complicaciones asociadas a la administración de la Alfa-Galactosidasa, únicamente las propias de las venoclisis (dolor, hematomas). DISCUSIÓN Y CONCLUSIONES • El enfermo afectado del Síndrome de Fabry es un enfermo crónico, con un tratamiento que no logra la recuperación integral pero que sí mejora su calidad de vida. AGRADECIMIENTOS Nuestro especial agradecimiento por su orientación, entusiasmo y paciencia en la realización de este trabajo a Naciba Zetchi y al Dr. Jordi Ara del Rey. BIBLIOGRAFÍA 1. Gupta S, Ries M, Kotsopoulos S, Shiffmann. The relationship of vascular Glycolipid Storage to Clinical Manifestations of Fabry Disease. A Cross- Sectional Study of a Large Cohort of Clinically Affected Heterozygous Woman. Medicine.2005; 84:5. | ||||||